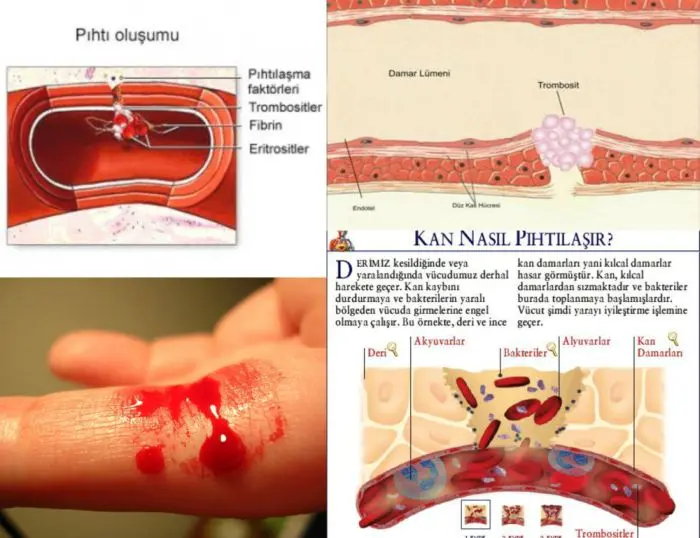
Gün içinde çeşitli sebeplerle yaralanabiliriz. Bu yaralanmalar bazen büyük bir kanamaya sebep olabilir. Kanama durumunda vücudumuzda bulunan pıhtılaşmadan sorumlu olan proteinler kanamanın durması ve yaranın iyileşmesi için devreye girerler. Bu durum bazı mutasyonlar sebebiyle gerçekleşemeyebilir ve kan pıhtılaşamaz.
Vücudumuzda oluşan bir yaranın kapanması için kanın pıhtılaşması gerekir. Vücudun kendi kendini onarabilmesi, bir kanamayı önlemesi veya herhangi bir nedenle başlayan kanamayı durdurması için oluşturduğu biyolojik süreçlere hemostaz denir. Hemostaz, yaralı damarda kan akışını durdurmanın bir yoludur. Hemostazın en önemli kısımlarından biri kanın pıhtılaşmasıdır. Hemostaz kan damarlarının daralması, trombositlerin aktivitesi ve kanda bulunan proteinlerin aktivitesi (pıhtılaşma faktörleri) olmak üzere üç ana süreçten oluşur. Yaralanmanın ardından ilk olarak kan kaybını kontrol etmek için kan damarı daralır ve etkilenen bölgede kan akışı sınırlandırılır. Yaranın çevresinden kolajen salgılanmaya başlar ve böylece trombositler aktif hale geçerek yaranın çevresine tutunur. Bu esnada kan pıhtılaşma faktörleri sırasıyla birbirlerini aktifleştirir. En son aktifleşen faktör 10 fibrin üretimini sağlar. İplik şeklindeki fibrinler trombositleri sıkılaştırır ve pıhtıyı güçlendirir. Yaranın boyutuna göre günler veya haftalar içinde kan damarı duvarı iyileşir ve zamanla kapanır.
Hemofili, pıhtılaşma faktörlerini üreten genlerdeki mutasyonlardan kaynaklanır. Pıhtı oluşumundaki bozukluğun nedeni karaciğerde üretilen faktör 8 (F8) ve faktör 9 (F9) proteinlerinin genetik hata yani bir DNA hatası nedeniyle fonksiyonunu yetersiz göstermesidir. Hemofili hastalığı kalıtsal bir bozukluktur. Ebeveynlerde bulunan genetik bozukluk çocuklara geçebilir. Bu mutasyonlar eşey hücrelerinden X'e bağlı resesif bir şekilde aktarılır. Erkekler XY, kadınlar XX kromozomlarına sahiptirler. Bu yüzden erkekler ya hasta ya da sağlıklı olurken kadınlar bütün bunlara ek olarak semptom göstermeden taşıyıcı da olabilir. Taşıyıcı oldukları durumda, kusurlu gen çocuklarına geçebilir ve gelecek nesilleri etkileyebilirler. Hastalığın erkeklerde görülme oranı kadınlarda görülme oranından daha fazladır.
F8 veya F9 genlerindeki mutasyonlar, pıhtılaşma faktörlerinin normal üretimini veya işlevselliğini bozar. Hemofili A'da mutasyonlar DNA dizisinde küçük değişikliklerden büyük silme (delesyon) veya tersine çevirmelere (inversiyon) kadar değişebilir ve hepsi Faktör 8'in yapısını ve üretimini etkiler. Benzer şekilde hemofili B, Faktör 9'un eksikliğine veya işlev bozukluğuna yol açan mutasyonlar tarafından oluşturulur.
Bir insanın kan plazmasının her 100 mililitresinde %50-150 ünite arasında pıhtılaşma faktörü bulunur. Bu düzeyin %40'ın altında indiği durum hemofili hastalığı olarak değerlendirilir. Bu düzey %5-%40 arasında ise hafif tip hemofili hastalığı denir. Bu düzeyin %1-%5 arasında olduğu durumlara orta tip hemofili denir.
Hemofili A yaklaşık on bin doğumda bir olarak görülürken, sıklığı Hemofili B'ye göre beş altı kat daha fazladır. Tüm hemofili olgularının %85'i Hemofili A, %15 kadarı ise hemofili B tipidir. Herhangi bir popülasyonda veya ırkta görülme sıklığı değişmeyen bu hastalık erkeklerde daha fazla görülmektedir.
Genetik araştırmalarındaki gelişmeler, hemofilinin genetik nedenlerine ilişkin anlayışımızı önemli ölçüde değiştirdi. DNA dizileme ve gen ekspresyon analizi gibi moleküler teknikler, bilim insanlarının hastalıkla ilişkili mutasyonları tanımlamalarını sağlamıştır. Bu gelişmeler, risk altındaki bireylerin belirlenmesine veya hemofili teşhislerinin doğrulanmasına yardımcı olan genetik test ve taşıyıcı taraması dâhil olmak üzere gelişmiş teşhis tekniklerinin yolunu açmıştır.
Genetik test ile genetik danışmanlık, aile planlaması ve kişiselleştirilmiş tedavi stratejileri için önemli bilgiler sağlanarak F8 veya F9 genlerindeki mutasyonlar tanımlanabilir. Ek olarak taşıyıcı taraması ile hemofili taşıyıcılarının saptanması, hastalığın gelecek nesillere geçme riskini azaltılabilir.
Şu anda hemofili için belirli bir tedavi yöntemi olmamasına rağmen bilimsel gelişmeler sayesinde herhangi bir yaralanma durumunda uygulanacak tedavi seçeneklerinde önemli ölçüde yol kat edilmiştir. Geleneksel tedavi, eksik pıhtılaşma faktörlerinin damardan uygulanarak bozuk faktörlerle değiştirilmesini içeriyordu.
Geleneksel yöntemin yanında son atılımlar gen terapisi gibi yenilikçi bir yaklaşımı da ortaya çıkarmıştır. Gen terapisi, normal pıhtılaşma faktörlerinin üretimine izin vererek mutasyona uğramış genlerin fonksiyonel kopyalarını hastanın vücuduna iletmeyi amaçlamaktadır. Bu gelecek vaat eden alan, klinik deneylerde önemli ilerlemeler göstererek uzun vadeli tedavi seçenekleri ve potansiyel tedaviler için umut vericidir. Aralık 2020'de Amerika Birleşik Devletleri Gıda ve İlaç Dairesi (FDA), hemofili A için ilk gen terapisi olan valoctocogene roxaparvovec (Valrox) 'u onaylamıştır.
Ayrıca CRISPR-Cas9 gibi gen düzenleme teknolojilerini kullanarak F8 ve F9 genlerindeki spesifik mutasyonları hedeflemeye ve düzeltmeye yönelik yöntemleri keşfetme çalışmaları devam etmektedir. Bu yaklaşım, hemofilinin temel nedenini potansiyel olarak ortadan kaldıran hassas genetik müdahaleler için büyük bir potansiyele sahiptir.
Hemofilinin genetik nedenleri bilimsel araştırmalar yoluyla çözümlenmiş ve bu nadir kanama bozukluğunun karmaşık yapısı anlaşılmıştır. F8 ve F9 genlerindeki mutasyonların anlaşılması tanısal teknikleri, genetik danışmanlığı ve kişiselleştirilmiş tedavi stratejilerini ilerletmiştir. Gen terapisi ve gen düzenleme teknolojilerindeki devam eden ilerlemeler, tedavi seçeneklerinin ve potansiyel tedavilerin geliştirilmesi için umut vaat etmektedir.